A comprehensive benchmark of single-cell Hi-C embedding tools
Published in Nature Communications, 2025
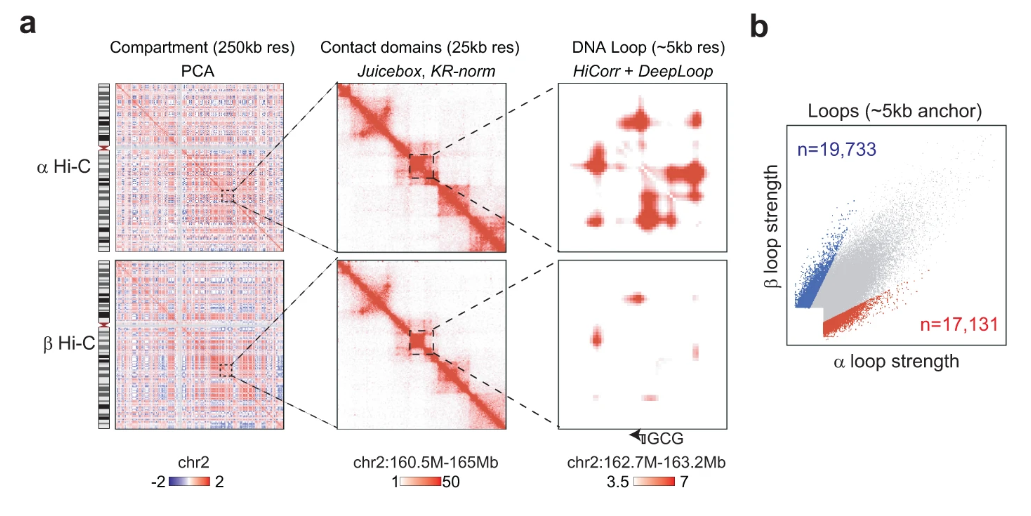
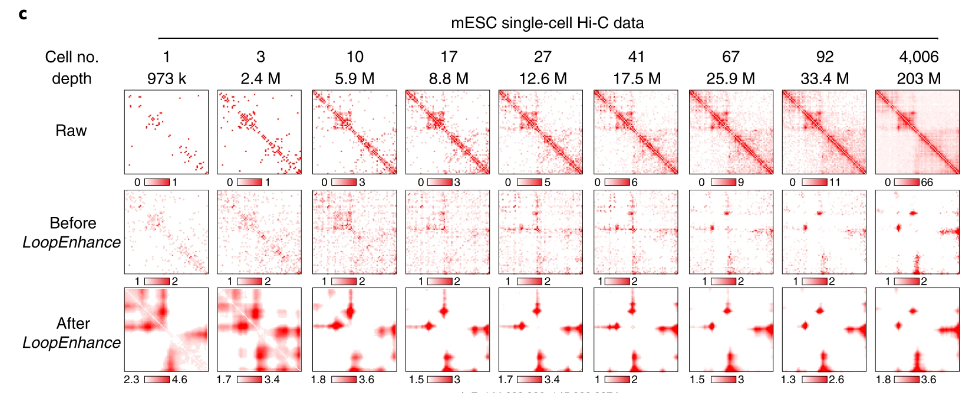
Embedding is the key step in single-cell Hi-C (scHi-C) analysis which relies on capturing biological meaningful heterogeneity at various levels of genome architecture. To understand the strength and limitations of existing tools in various applications, here we use ten scHi-C datasets to benchmark thirteen embedding tools including Va3DE, a new convolutional neural network model that can accommodate large cell numbers. We built a software framework to decouple the preprocessing options of existing tools and found that no single tool works best across all datasets under default settings. The difficulty levels and preferred resolutions are different between benchmark datasets, and the choice of data representation and preprocessing strongly impact the embedding performance. Embedding cells from early embryonic stages relies on long-range compartment-scale contacts, but resolving cell cycle phases and complex tissue requires short-range loop-scale contacts. Both random-walk and inverse document frequency (IDF) transformation prefers long-range “compartment-scale” over short-range “loop-scale” embedding, while deep-learning methods better overcome sparsity at both scales and are more versatile with different resolutions. Finally, “diagonal integration” with independent data modal is a promising approach to distinguish similar cell subpopulations. Our findings underscore the significance of appropriate priors for scHi-C embedding and also offer insights into genome architecture heterogeneity. 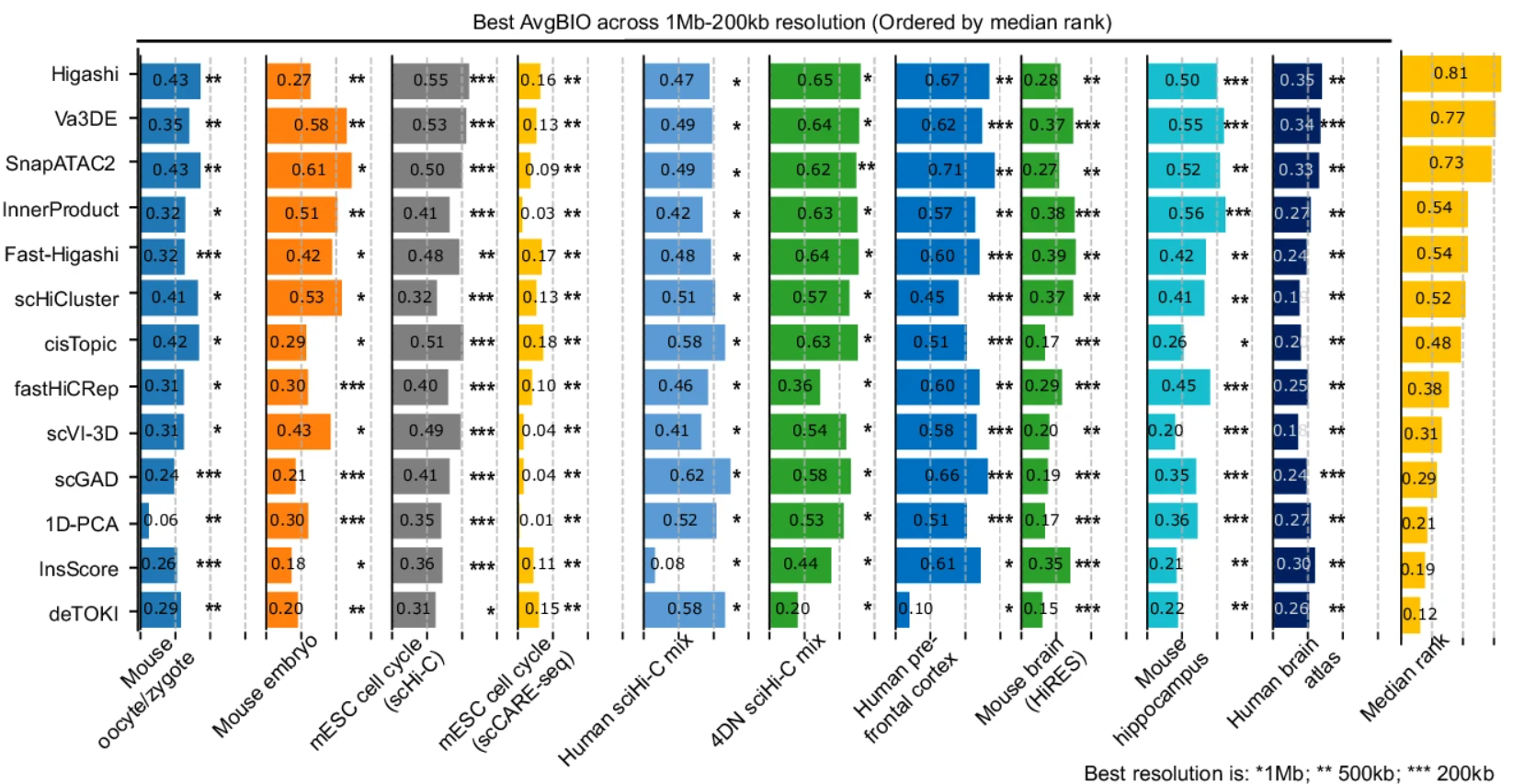
Recommended citation: Plummer, D., Lang, X., Zhang, S. et al. A comprehensive benchmark of single-cell Hi-C embedding tools. Nat Commun 16, 9119 (2025). https://doi.org/10.1038/s41467-025-64186-4 https://www.nature.com/articles/s41467-025-64186-4