
DeepLoop robustly maps chromatin interactions from sparse allele-resolved or single-cell Hi-C data at kilobase resolution
Published in Nature Genetics, 2022
Recommended citation: Zhang, S., Plummer, D., Lu, L. et al. DeepLoop robustly maps chromatin interactions from sparse allele-resolved or single-cell Hi-C data at kilobase resolution. Nat Genet 54, 1013–1025 (2022). https://doi.org/10.1038/s41588-022-01116-w https://www.nature.com/articles/s41588-022-01116-w
In this work I trained some simple convolutional autoencoders with surprisingly effective results. Our main observations are that by rigorously removing as many sources of bias as possible using HiCorr, we can train a single denoising autoencoder to remove noise from high resolution Hi-C from any technological platform (e.g conventional Hi-C, in situ Hi-C, Micro-C, etc). After verifying the consistency of the model outputs across platforms and species, we can then train a series of deeper models to reconstruct these clean loop signals from very low-depth data. One of the main benefits of this is lowering the barrier to high resolution loop-level analysis to only ~10M cis-reads, thereby democratizing Hi-C studies and allowing loop-level retro-active analysis of existing datasets. We mainly highlight two of the exciting possible use-cases for such a model:
High resolution loop-level analysis of single-cell Hi-C
Allele-specific loop analysis
Single-cell Hi-C is able to map chromatin contacts in individual cells. However, each cell typically has <1M total reads, which is entirely insufficient for high resolution analysis. We find though, that with DeepLoop, we can predict reproducible loop signals from only a dozen cells:
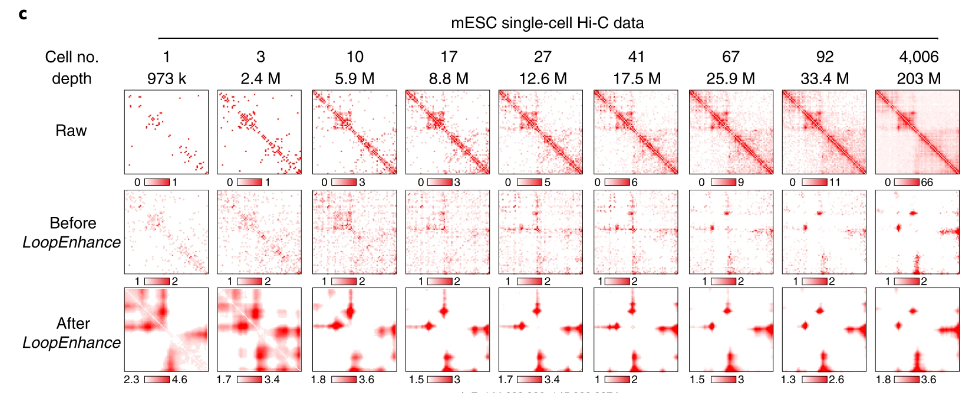
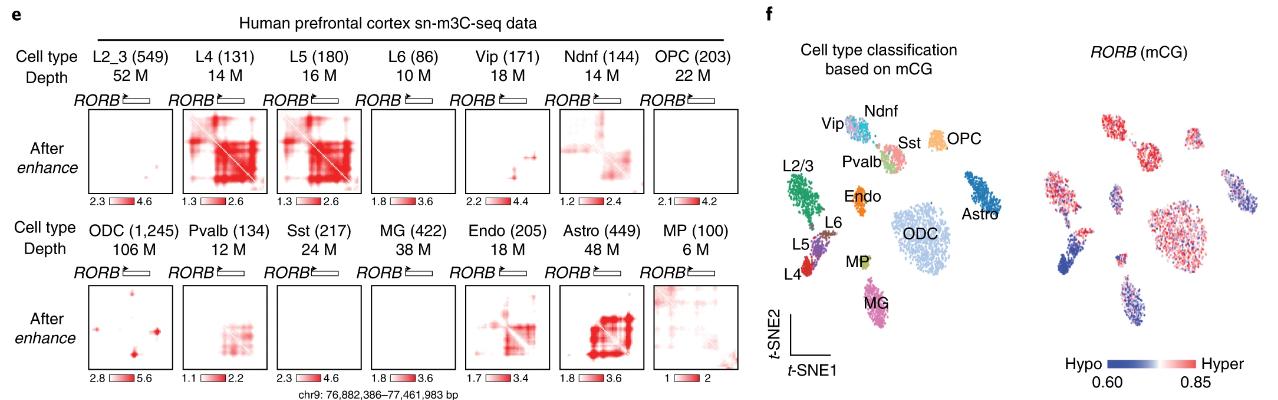
This is particularly exciting from the perspective of single-cell Hi-C clustering. If we can identify cell-type specific clusters with a minimum of around a dozen cells, we can create pseudo-bulk datasets which are sufficient for DeepLoop analysis. We test this in a co-assayed dataset profiling both chromatin contacts and DNA methylation in human prefrontal cortex cells. We choose this paired data because we can be confident in the cluster assignments from DNA methylation embedding. Embedding and clustering of chromatin contacts is still very much an active area of research. We find that the loop-specific heatmaps align well with hyper and hypomethylated loci in difference celltypes:
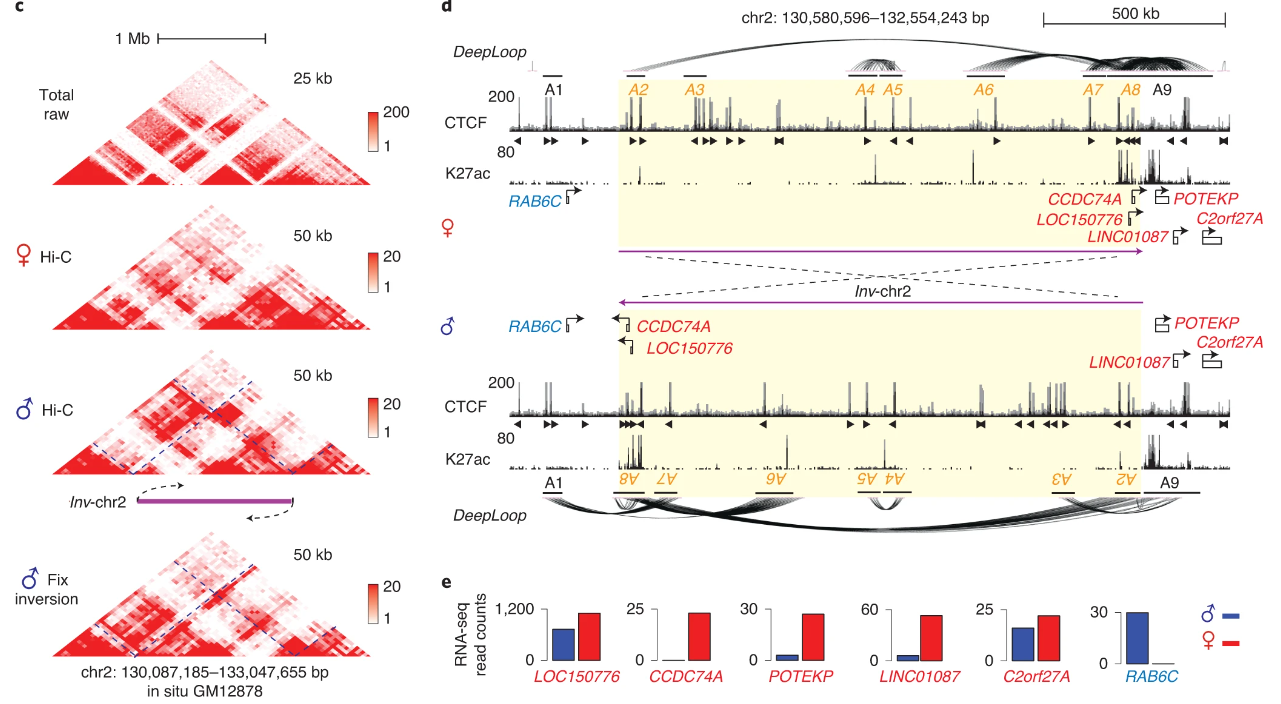
The other most exciting application of DeepLoop that we demonstrate is the ability to analyze allele-specific loops. We start with a billion-reads dataset. Of these 4.5 billion reads, we can find ~300M which overlap with known SNPs, so we can map each read to its corresponding allele. This gives us two new low-depth datasets which previously could not be analyzed at the loop level.
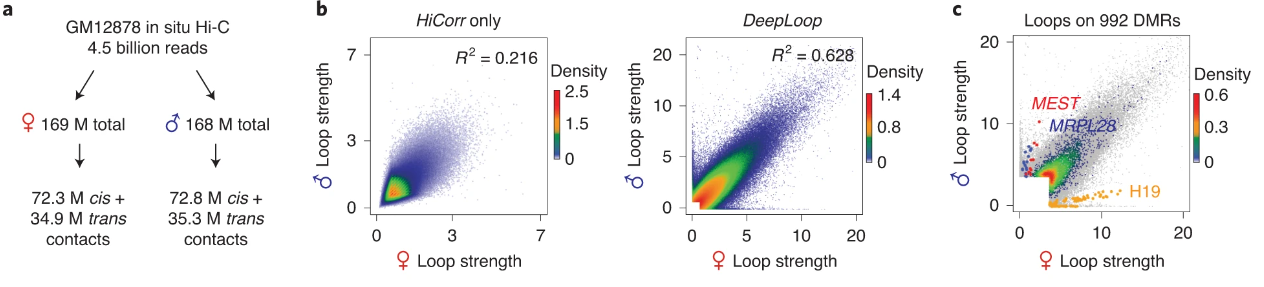
Excitingly, since we only need ~10-20M cis-reads to confidently apply DeepLoop, we can analyze allele-resolved Hi-C at unprecedented resolution revealing homolog-specific chromatin loops.
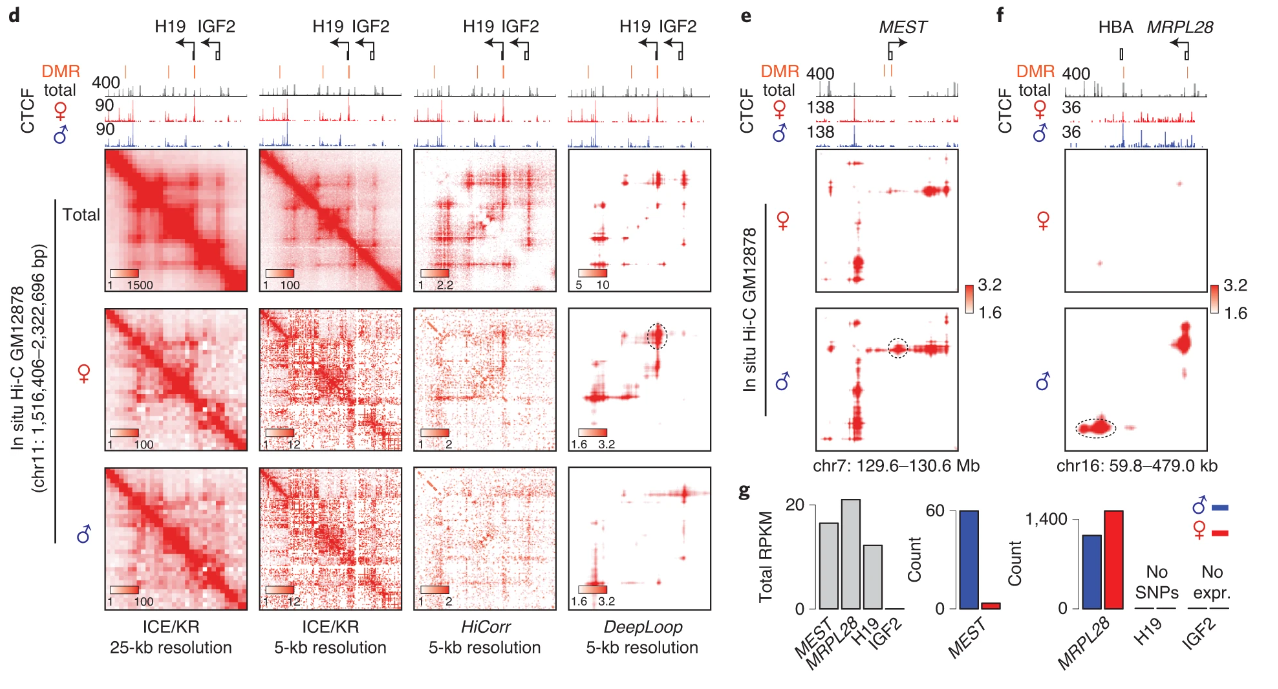
By isolating the extremely allele-specific interactions, we can also pinpoint heterozygous SNVs. Such SNVs were missed by previous studies because of low resolution (1Mb) and the assumption of a homozygous genome.

There are lots of other exciting applications of these models explored in the paper! Overall we hope that these models and this perspective on bias-corrected Hi-C will be widely adopted by the field. The pipeline has already become a staple in the Jin Lab’s Hi-C analysis pipeline and we hope that it will continue to uncover interesting biology in the structure of the 3D genome.
Below is an interactive example of some specific structures in H1 vs GM12878 visualized with a force-directed graph layout: