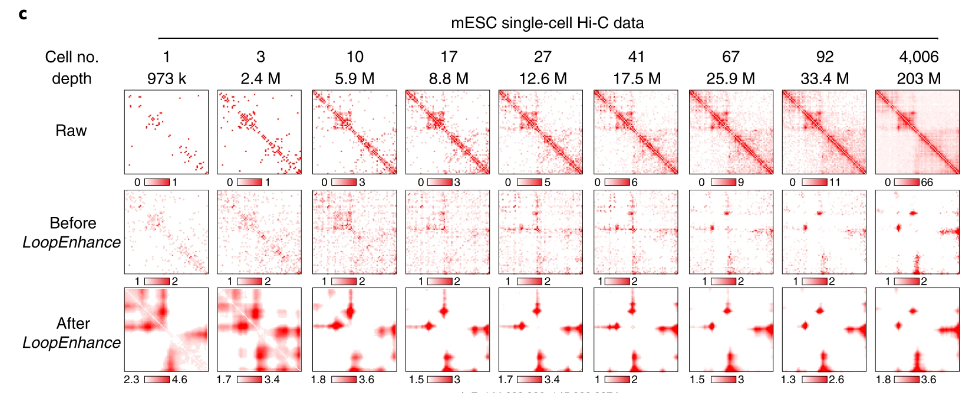
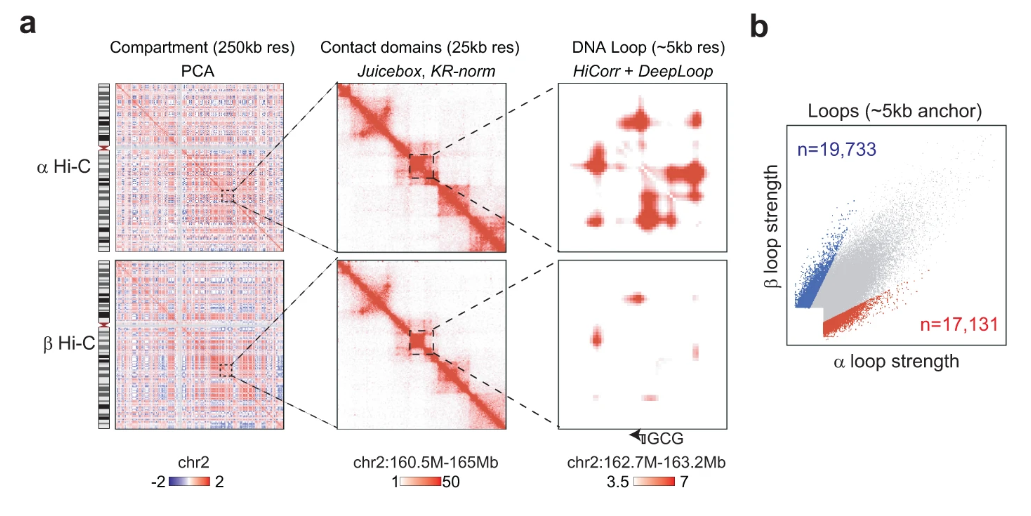
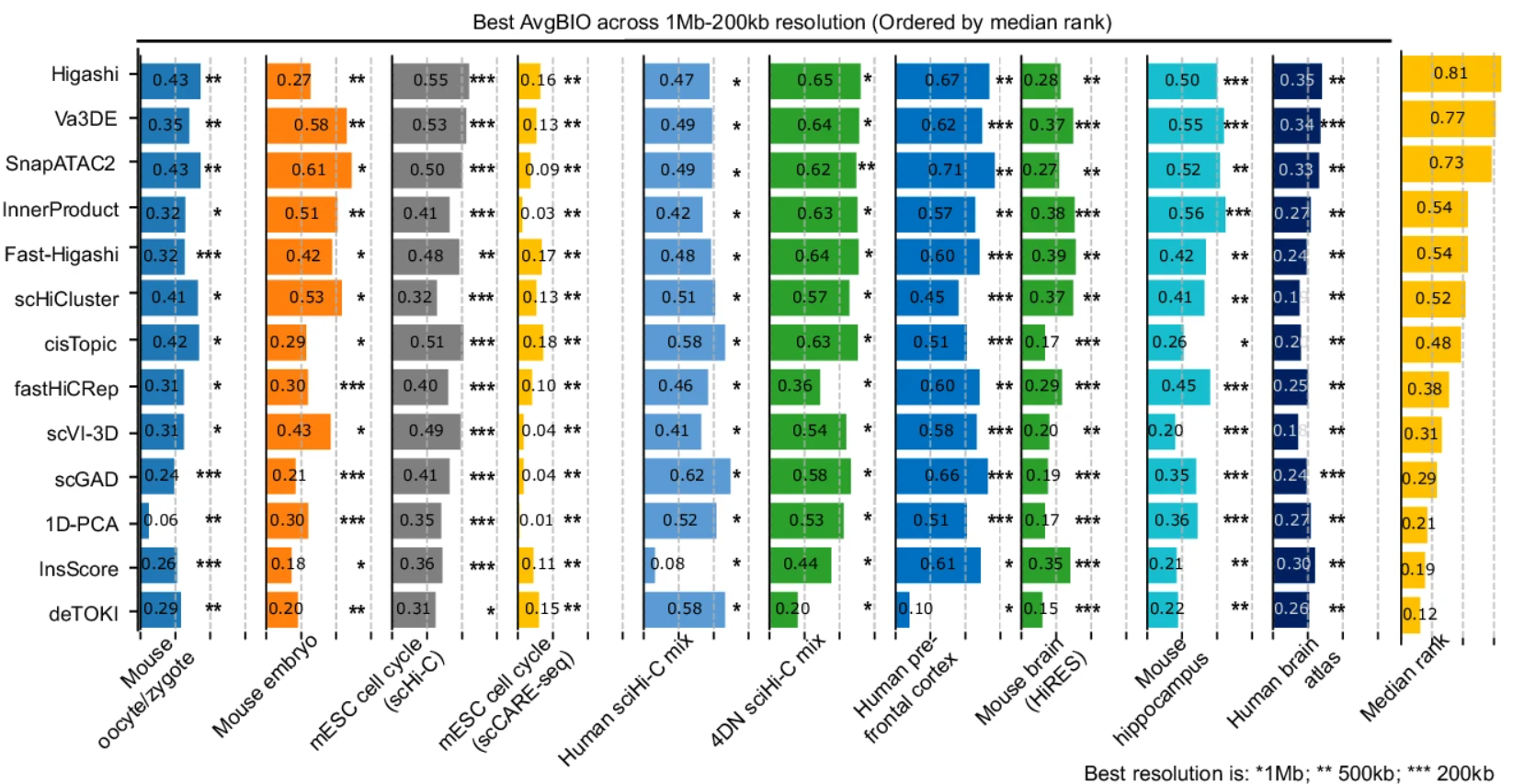
Published in Case Western Reserve University ProQuest Dissertations & Theses, 2024
Genome sequencing has unlocked the ability to understand the genetic basis of human disease and development. However, the genome is not a linear sequence of bases, but a complex 3D structure of interacting regulatory elements. This structure is dynamic and varies between celltypes. By leveraging Hi-C data—a powerful tool for studying the 3D genome—we can begin to understand the interactions that regulate gene expression. But Hi-C data is high dimensional, noisy, and sparse; and the 3D genome is difficult to interpret in relation to other genomic data. To address this, we take a representation learning approach to the 3D genome. We introduce a set of autoencoding models: DeepLoop for bulk data, Va3DE for single-cell data, and HiGLUE for multi-modal learning. We demonstrate that simply training neural networks to compress and reconstruct chromatin loop information from Hi-C datasets can lead to the discovery of biologically relevant features. We produce some of the first ever allele-specific chromatin loop maps as well as the first Hi-C maps of celltypes in the pancreatic islet through multimodal integration of single-cell Hi-C data. After learning a representation of the 3D genome and its relationship to other genomic data, we can then simulate the effects of genetic perturbations and verify regulatory relationships or identify disease pathways. Due to the newly formed symbiotic data-driven relationship between genomics and machine learning, our field will continue developing and iterating upon these various models in tandem with new protocols for generating data, eventually converging on comprehensive models of the dynamic genome which can be queried, perturbed, and simulated in larger biological systems. We present a comprehensive exploration into how the 3D genome should be included in this new generation of research.
Recommended citation: Plummer, D. Loop2Loop: Representation Learning of the 3D Genome for Multimodal Single-Cell Integration and In-Silico Chromatin Rewiring. Doctoral dissertation, Case Western Reserve University. (2024) https://www.proquest.com/openview/c814f710061a3c2a4caddc5b9a8e012c/