
A comprehensive benchmark of single-cell Hi-C embedding tools
Published in Nature Communications, 2025
Recommended citation: Plummer, D., Lang, X., Zhang, S. et al. A comprehensive benchmark of single-cell Hi-C embedding tools. Nat Commun 16, 9119 (2025). https://doi.org/10.1038/s41467-025-64186-4 https://www.nature.com/articles/s41467-025-64186-4
In this work we perform the first comprehensive benchmark of single-cell Hi-C (scHi-C) embedding tools, evaluating thirteen methods across ten diverse datasets. We also introduce Va3DE, a new variational convolutional neural network model designed to scale to large cell numbers.
A key contribution of this study is the development of a modular software framework that decouples the preprocessing options from the embedding methods themselves. This allowed us to systematically evaluate how different choices in data representation and preprocessing impact downstream embedding quality — often more so than the choice of embedding algorithm itself.
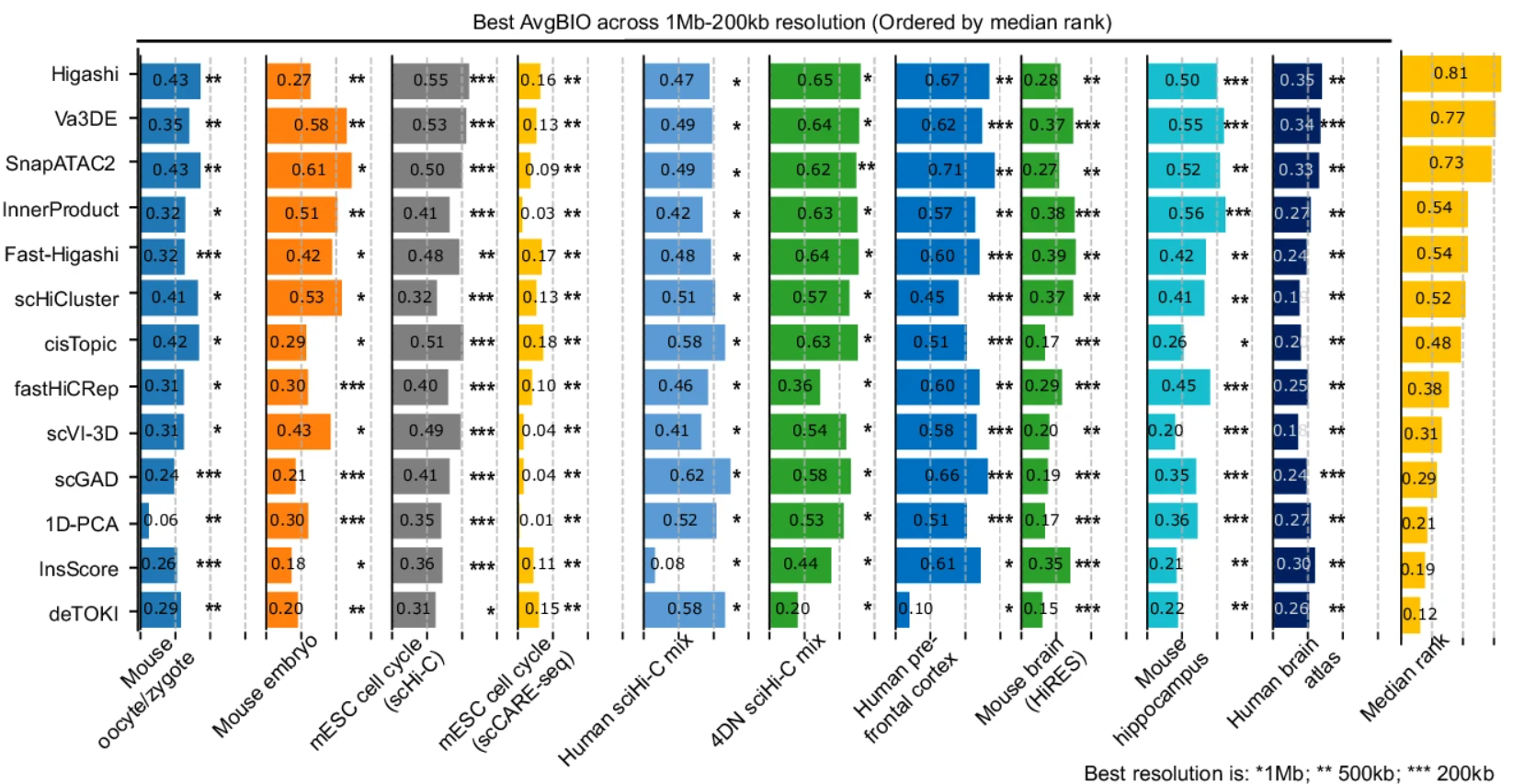
Our main findings reveal that no single tool works best across all datasets under default settings. The difficulty levels and preferred resolutions vary substantially between benchmark datasets, highlighting the importance of matching analysis parameters to the biology of interest:
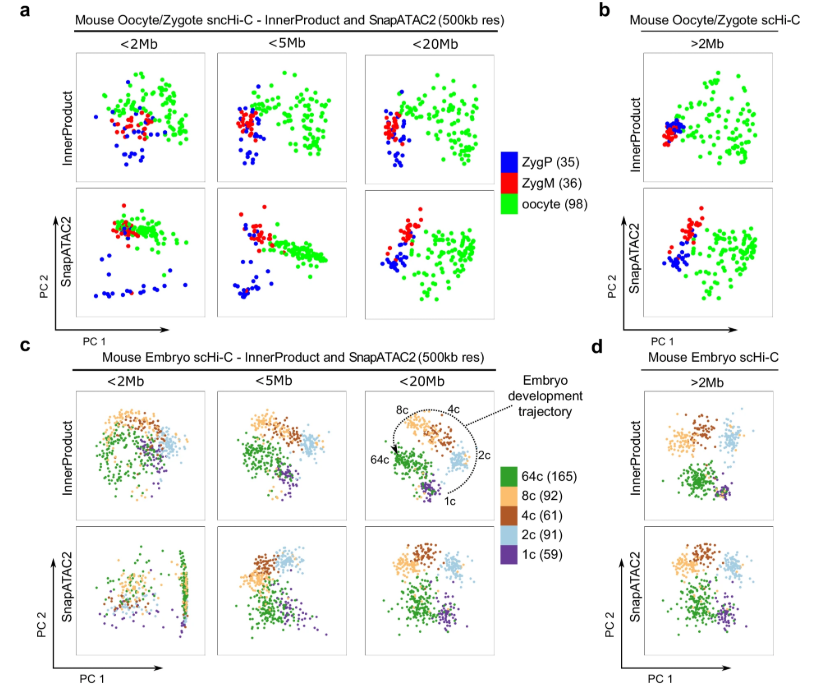
- Compartment-scale contacts (long-range) are essential for resolving cells from early embryonic stages, where large-scale genome reorganization drives cellular identity.
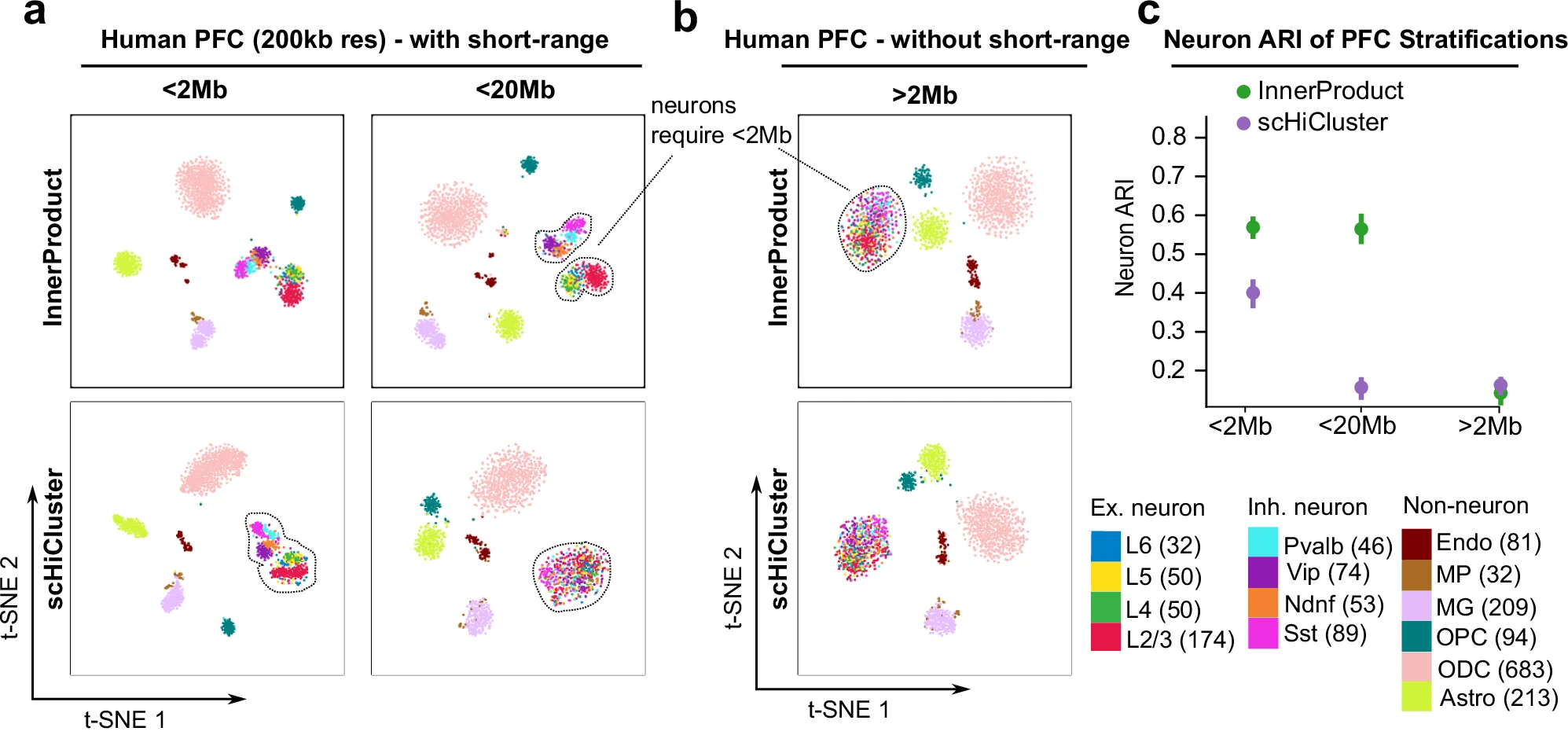
- Loop-scale contacts (short-range) are critical for distinguishing cell cycle phases and resolving complex tissue heterogeneity, where fine-grained regulatory differences matter.
We also find important distinctions between preprocessing strategies. Both random-walk-based and inverse document frequency (IDF) transformations tend to favor long-range “compartment-scale” embedding, while deep-learning-based methods are more versatile — they better overcome the extreme sparsity of scHi-C data at both compartment and loop scales and are more robust across different resolutions.
Finally, we explore “diagonal integration,” an approach that combines independent data modalities along the contact distance axis. This strategy shows particular promise for distinguishing closely related cell subpopulations that are difficult to separate using any single scale of genome architecture alone.
Overall, our findings underscore that appropriate priors — including resolution, distance range, and preprocessing — are critical for effective scHi-C embedding. We hope this benchmark and the accompanying framework will serve as a practical guide for the community and help researchers make informed choices when analyzing single-cell chromatin conformation data.